研究トピックスResearch Topics
アミノ酸バランスを活用した治療抵抗性克服:“抗がん剤ブースター療法”の開発Amino acid imbalance overcomes therapy resistance in brain tumor
Jing Yongwei, Masahiko Kobayashi et al., Nature Communications 2025
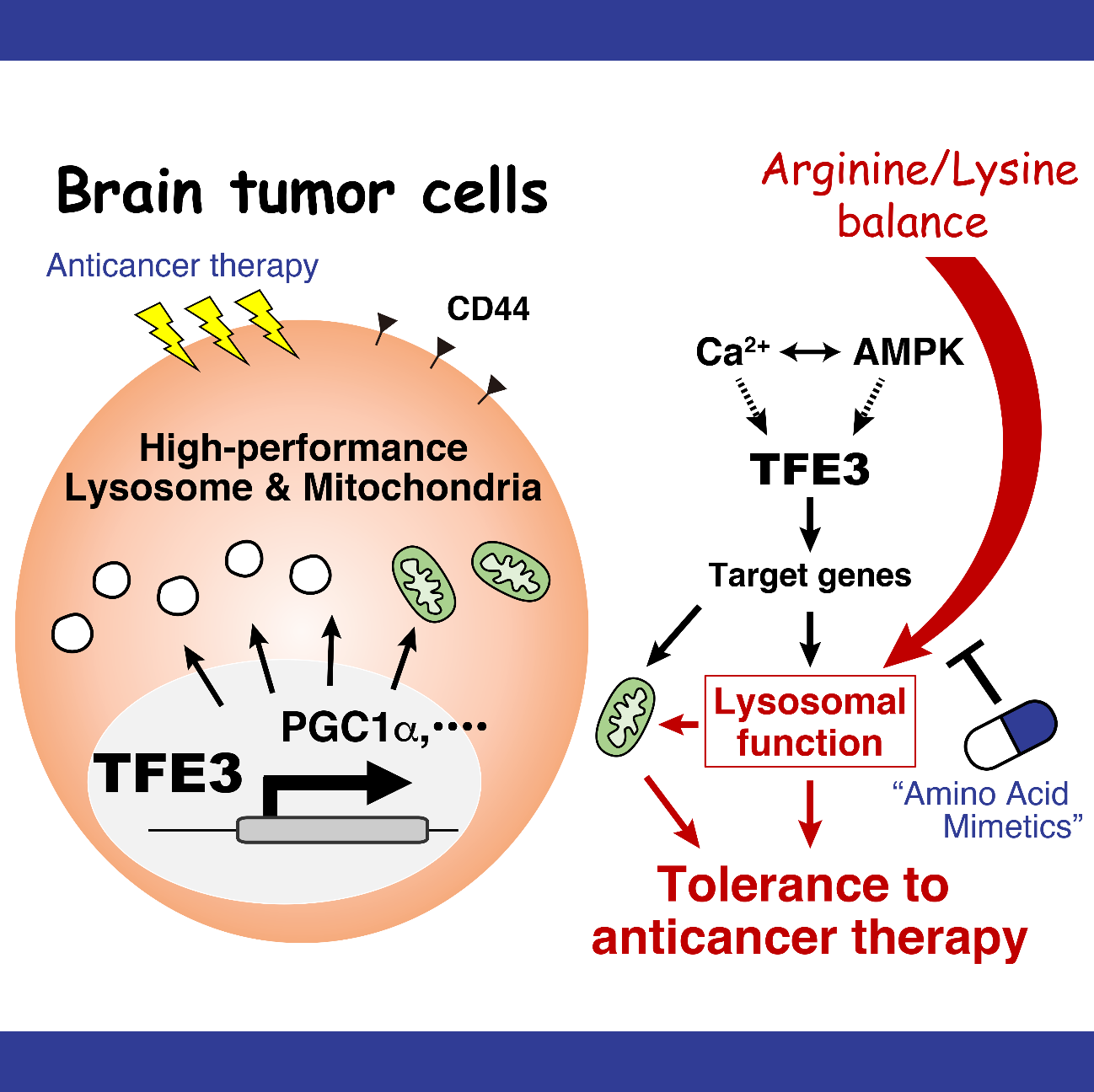
悪性膠芽腫患者由来細胞株の解析により、これらの細胞は正常な神経前駆細胞に比べてリソソームの活性が高いこと、リソソーム活性が高い腫瘍細胞ほど、酸素を利用するミトコンドリアの活動が活発であり、特徴的な代謝状態を持つことが確認された。そこで、リソソーム活性を制御する因子を調べたところ、リソソームの生合成を調節するタンパク質「TFE3」が治療耐性に深く関与していることが判明した。TFE3は、PGC1αなどの分子を介して腫瘍細胞の特異的な代謝を調整していることも明らかになった。
これらの結果をもとに、腫瘍細胞の代謝をさらに詳しく解析したところ、アミノ酸の動きに大きな変化が見られた。そこで、培養液からアミノ酸を一つずつ除去して細胞を培養し、リソソーム活性への影響を調べた結果、「リシン」がリソソームの機能維持に不可欠であることを見いだした。培養液中のリシン濃度を低下させると、リソソームの機能が低下し、抗がん剤「テモゾロミド」に対する腫瘍細胞の感受性(治療効果)が高まることも確認された。そのメカニズムとして、リシンがアルギニンを利用した一酸化窒素の生成を抑えることで、腫瘍細胞をリソソームストレスから保護する役割を果たしていることが明らかになった。
リシンは体内で合成できないため、食事から摂取する必要がある「必須アミノ酸」の一つである。例えば、リシンを含まない特別食により体内のリシンを制限すると、体重が大きく減少する。そこで、リシン制限と同様の効果を持つ化合物を探索した結果、「ホモアルギニン」がテモゾロミドの治療効果を高めることを発見した。ヒト脳腫瘍細胞を免疫不全マウスに移植したモデルを用いた実験では、ホモアルギニン単独では腫瘍に対する顕著な効果は見られなかったが、テモゾロミドと併用することで抗腫瘍効果が増強され、マウスの生存期間が延長することが確認された。以上の結果から、ホモアルギニンの投与は、テモゾロミドの治療効果を高める「抗がん剤ブースター療法」として有望であることが示された。
Analysis of patient-derived malignant glioblastoma cell lines revealed that these cells exhibit higher lysosomal activity compared to normal neural progenitor cells. Furthermore, tumor cells with higher lysosomal activity showed increased mitochondrial activity, particularly in oxygen-dependent metabolism, indicating a unique metabolic state. Next, we investigated factors regulating lysosomal activity and identified the transcription factor TFE3, which regulates lysosomal biogenesis, as a key contributor to therapeutic resistance. We also found that TFE3 modulates the characteristic metabolism of tumor cells through molecules such as PGC1α.
Based on these findings, we conducted a detailed metabolic analysis of tumor cells and observed significant alterations in amino acid dynamics. To explore this further, we systematically removed individual amino acids from the culture medium and examined the effects on lysosomal activity. This experiment identified lysine as an essential factor for maintaining lysosomal function. Furthermore, reducing lysine levels in the culture medium led to impaired lysosomal function and increased tumor cell sensitivity to the chemotherapeutic agent temozolomide, thereby enhancing its therapeutic efficacy. Mechanistically, we demonstrated that lysine plays a protective role in tumor cells by suppressing nitric oxide production via arginine utilization, thereby mitigating lysosomal stress.
Lysine is an essential amino acid that cannot be synthesized endogenously and must be obtained through dietary intake. For instance, dietary lysine restriction through a lysine-deficient diet is known to cause significant body weight loss. To achieve a similar effect without dietary intervention, we screened for compounds that mimic lysine restriction and identified homoarginine as a potential enhancer of temozolomide efficacy. In an immunodeficient mouse model transplanted with human glioblastoma cells, homoarginine alone did not exert a significant antitumor effect. However, when combined with temozolomide, it significantly enhanced the therapeutic response and prolonged the survival of tumor-bearing mice.
Collectively, these findings suggest that homoarginine administration serves as an effective "chemotherapeutic booster" strategy, augmenting the efficacy of temozolomide in glioblastoma treatment.
高脂肪食による全身の代謝異常状態から造血幹細胞を守るしくみ,発見!Mechanism to safeguard hematopoietic stem cells against excessively unbalanced diet
Yuko Tadokoro et al., Cell Stem Cell 22, 1-13 (2018)
造血幹細胞は,全ての血液細胞を供給する血液の源の細胞として,全ての血液細胞を産生する能力(多分化能)と共に,自身が失われないように自己を複製する能力(自己複製能)を兼ね備えている。私たちの体は,日々,炎症や活性酸素などさまざまなストレスにさらされているが,一生涯にわたって血液細胞を供給し続けるためには,これらのストレスから造血幹細胞を守るシステムが必要である。最近,極端に偏った食生活,特に脂肪分の多い食事は,生活習慣病やがんなどさまざまな疾患の要因になることが知られているが,このような極端に偏った食事が原因となるストレスが,造血幹細胞にどのような影響を与えているのかは,これまで明らかにはされてこなかった。
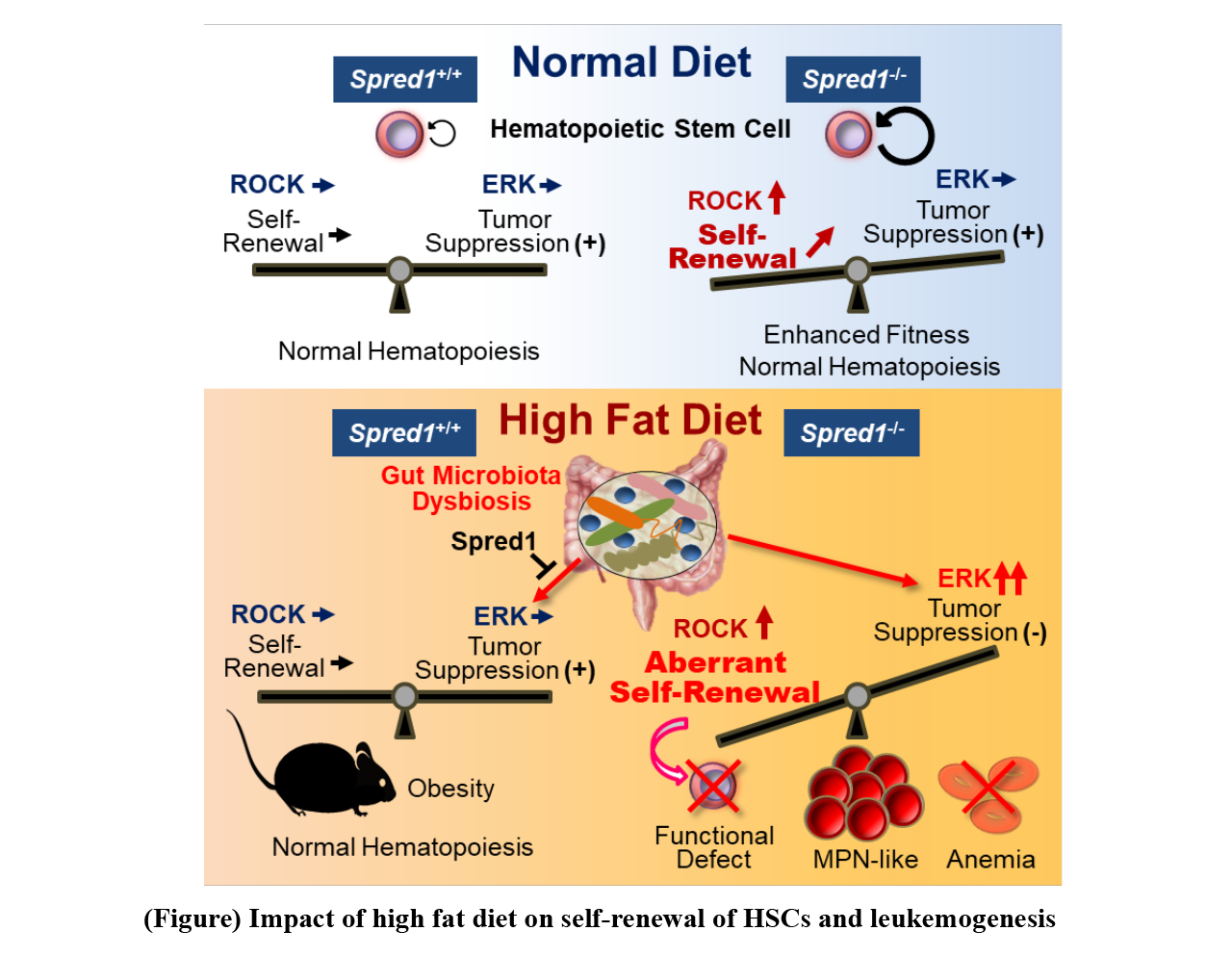
我々は,これまで,造血幹細胞の自己複製に関わる分子やシグナルを研究し,その過程において,造血幹細胞の自己複製能はSpred1という分子によるROCKシグナルの制御によって調節されていることを発見した。さらに,このSpred1は造血幹細胞において,サイトカイン,加齢,放射線障害,細菌感染などの刺激によって活性化することを見いだした。そこで,Spred1を欠損するマウスを用いて,造血幹細胞におけるSpred1の役割を調べた結果,Spred1欠損マウスは通常の飼育条件では特に顕著な血液の異常を示さなかったが,上記のストレスを与えた際,野生型マウスでは造血幹細胞の傷害が観察されるのに対して,その傷害は軽減される,あるいは,通常より造血幹細胞の機能が亢進することが判明した(図)。このことから,Spred1は,造血幹細胞の自己複製を抑える機能を有するものの,通常の血液の産生において重要な役割を果たしているわけではないと考えられた。
次に,さまざまなストレスを負荷する実験を進めた結果,ラードを多く含む高脂肪食を与え続けると,Spred1欠損マウスは白血病を発症し,死亡することを観察した。この時,高脂肪食を与えたSpred1欠損マウスの造血幹細胞では,その正常な機能が失われると同時に,ERKシグナルが異常に活性化し,白血病を引き起こすことを見いだした。このことから,Spred1は過剰な脂肪摂取によるがん化シグナルの活性化を抑制し,過栄養ストレスに対して造血幹細胞の機能を正常に保つ働きがあると考えられた。さらに我々は,Spred1欠損マウスにおいて,過剰な脂肪摂取が白血病発症を誘導する原因について調べた。その結果,高脂肪食を与えたマウスでは通常食を与えたマウスに比べて腸内でのグラム陽性菌の割合が増えており,腸内細菌叢のバランスが崩れていることを見いだした。そこで,高脂肪食を与えたSpred1欠損マウスに抗生物質を投与して腸内の除菌を行ったところ,造血幹細胞の機能低下や白血病の発症が抑制された。以上のことから,高脂肪の食事は腸内細菌叢の変化を介して造血幹細胞の制御に影響を及ぼしていることに加え,通常ではSpred1がそのようなストレスから造血幹細胞が機能を失って白血病化しないように守っているが,そのシステムが破たんすると造血幹細胞に傷害を与え,白血病発症の原因になると考えられた(図)。
適切な食習慣を続けることは,健康を維持する上で最も重要な要素である。今回のマウスの実験では,たとえ,過剰な脂肪の摂取を続けても,通常は,Spred1が造血幹細胞の守護神として機能することによって,極端な血液異常の危険性を回避していると考えられた。
最近の研究で,一部の白血病患者ではSpred1の発現低下が見られ,しかも,そのような患者は,そうでない患者より重い病状を示すことも判明している。つまり,何らかの原因でSpred1の機能が弱くなっている場合,暴飲暴食を続けると,血液の異常が生じる可能性がある。さらに,研究を進めることによって,将来,白血病の予防や治療法の向上につながるものと期待される。
Hematopoietic stem cells (HSCs) can give rise to all types of blood cells during whole life of animals. HSCs possess multipotency to supply all types of blood cells as well as to reproduce themselves (self-renewal) in order not to be depleted. Since the body is always exposed to various kinds of stresses such as inflammation and reactive oxygen species, it is considered that a system is necessary to protect HSCs from various stresses in order to continuously supply all types of blood cells throughout life. Recently, it has been known that an excessively unbalanced diet, especially a high-fat diet, may be one of the important factors to cause various diseases including lifestyle related diseases and cancer. However, it was not known how stresses caused by an excessively unbalanced diet would affect HSCs.
We have been conducting research on molecules and signaling pathways involved in HSC self-renewal. The team found that the ability for self-renewal of HSCs was regulated by ROCK signaling controlled by the Spred1 molecule. Furthermore, the team discovered that Spred1 in HSCs was activated by stimuli like cytokines, aging, radiation damage, bacterial infection and so on. Here, roles of Spred1 in HSCs were investigated by using Spred1-deficient mice. Under normal conditions, Spred1-deficient mice did not show any significant disorder of the blood; however, under a condition with one of the stressors mentioned above, while wild type mice showed damage of HSCs, Spred1-deficient mice showed either reduced damage or augmentation of HSC functions (Figure). These results suggest that although Spred1 functions to suppress self-renewal of HSCs, it does not play important roles in HSCs’ giving rise to the blood cells under normal conditions. Next, by carrying out experiments under various forms of stress, the team has found that Spred1-deficient mice initiate leukemogenesis and die after having continuously been fed a high-fat diet containing large amounts of lard. With these Spred1-deficient mice, HSCs have been found to lose normal functions but their ERK signaling has been aberrantly activated to initiate leukemogenesis. These results indicate that Spred1 suppresses activation of tumorigenesis-stimulating signaling pathways induced by excessive fat intake and that Spred1 plays an important role in maintaining normal HSC functions against excessive nutritional stress.
In addition, we investigated the reasons why excessive fat intake initiates leukemogenesis in the Spred1-deficient mice. It has been found that the ratio of gram-positive bacteria in the gut is increased in the mice fed a high-fat diet in comparison with the mice fed a normal diet, equilibrium of the gut microbiota being disrupted. By administering antibiotics to the Spread1-deficient mice fed a high-fat diet to eliminate bacteria in the gut, suppression of reduction of HSC functions is observed along with suppression of initiation of leukemogenesis. With these results, the research team concludes that a high-fat diet affects regulation of HSCs by altering the gut microbiota. Under normal conditions, Spred1 protects HSCs from losing their activity against dietary stress-induced initiation of leukemogenesis, whereas a failure of the system damages HSCs, possibly initiating leukemogenesis (Figure).
A well-balanced and appropriate diet is one of the most important factors for maintaining good health. The present research using mice indicates that, even with continuous excessive fat meals, Spred1 plays an important role as a safeguard of HSCs under normal conditions to avoid serious abnormalities of the blood. Recent research has, however, revealed that some leukemia patients have low expression levels of Spred1 and that such patients suffer from severer pathology. Therefore, if Spred1 function is impaired for some reason, continuous overeating and overdrinking would possibly lead to abnormality of the blood. The team hopes to elucidate the pathophysiology of blood abnormalities such as leukemia, by investigating the regulation of expression level of Spred1 and gut microbiota-derived factors induced by excessively unbalanced dietary habits, and by further investigating regulatory mechanisms of Spred1 functions. We believe that further research will lead to prevention of leukemia and to improvement of therapy.
ニコチンアミド代謝物を定量できる超分子バイオセンサー!Supramolecular biosensors for a nicotinamide metabolite
Masaya Ueno et al, Comm. Chemistry (2020), Masaya Ueno, Hiroki Sugiyama et al, Anal. Chem (2024)
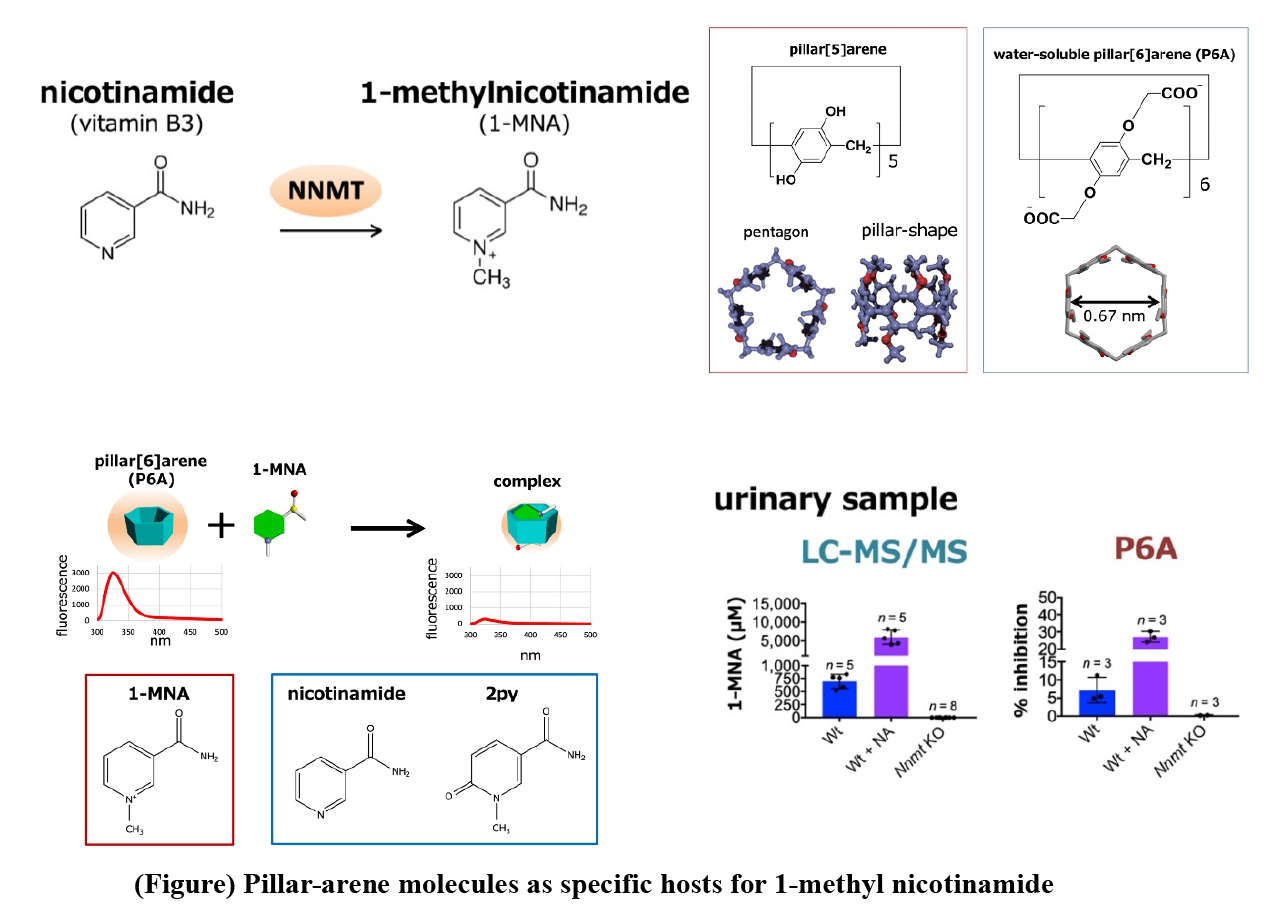
食物から取り込まれたアミノ酸,糖質,核酸,脂肪,ビタミンなどの栄養素は,体内のさまざまな酵素によって分解や化学的修飾を受けて代謝され,体内では非常に多種多様な代謝物が作り出される。それぞれの代謝過程は,栄養素の摂取量や排泄量,エネルギーの蓄積量や消費量などさまざまな生理状態に応じて綿密に制御されている。体内でニコチンアミドは,ニコチンアミド・アデニン・ジヌクレオチド(NAD+)の生合成に使われると同時に,代謝酵素NNMTによって1-メチルニコチンアミド(1-MNA)となる(図)。一部の1-MNAは,さらにアルデヒドオキシダーゼによって酸化され,2py(N1-メチル-2-ピリドン-5-カルボキサミド)あるいは4py(N1-メチル-4-ピリドン-3-カルボキサミド)となり,最終的に1-MNA,2py,および4pyはいずれも尿中に排泄される。一方で,最近の研究から,NNMTはさまざまながんで発現が亢進しており,がんの進展や悪性化にNNMTが関与していることが明らかになってきた。NNMTを抑制することで,いくつかのがん細胞株の増殖を抑制できることが報告されており,がん細胞のNNMTを阻害できれば,がんを抑制できる可能性がある。
今回,1-MNAの簡便な測定方法を開発する目的で,超分子化学(生越教授:京都大学)の研究者らと共同研究を行い,水溶性ピラー[6]アレーン(P6A)が1-MNAと特異的に結合し,この結合によりP6Aの蛍光が消光することを発見した。P6Aは生越教授らの研究グループによって最初に報告された超分子化合物“ピラーアレーン”が1-MNAのような分子サイズの陽イオン分子と結合するように設計した化合物で,1,4-ジアルコキシベンゼンがパラ位で結合した柱型環状化合物から合成される(図)。ピラーアレーンは分子中央の電子豊富な空洞を持ち,その空洞サイズの適した陽イオン分子と安定した錯体を形成することが知られている。我々はプロトン核磁気共鳴(1H NMR)および等温滴定型カロリメトリー(isothermal titration calorimetry)解析から,P6Aは1:1の割合で1-MNAと安定な錯体を形成することを発見した。P6Aの結合は高い選択性があり,化学構造が非常に類似しているニコチンアミドや2pyとはほとんど結合しないことを明らかにした(図)。さらに,NNMTの無細胞反応系や,Nnmt欠損マウスを用いた解析から,P6Aは夾雑物が含まれる酵素反応液や尿試料中でも1-MNAと特異的に結合し,1-MNA濃度に依存して蛍光が消光することを発見し,簡便に,質量分析計を用いた従来法と同等の結果が得られることを確認した(図)。
これらの知見から,がん患者の迅速診断に活用されることが期待される。さらに,がん細胞の表層や内部の代謝物を直接観ることができるナノプローブの開発や,がんの診断や治療に有用な全く新しいバイオセンサー開発への応用が期待される。
Metabolites are organic molecules that take part in or are created during the biochemical reactions constantly taking place in an organism. The presence of certain metabolites can be an indicator for particular pathological conditions related to cancers. Therefore, efficiently measuring and monitoring the presence is therefore important for early diagnosis. We have developed a biosensor for a low-molecular-weight metabolite known as 1-MNA. The sensor relies on the physicochemical properties of pillar[6]arene, a channel-like molecule. The first pillararene was synthesized in 2008 by Dr. Tomoki Ogoshi and colleagues from Kanazawa University. The name pillararene was chosen since the molecules are cylindrical (pillar-like) in shape and composed of aromatic moieties (arenes). We investigated the metabolite 1-MNA (1-methylnicotinamide), a product of NNMT (nicotinamide N-methyltransferase). Since it has been shown that NNMT activation causes malignant properties of cancers, detecting 1-MNA could be therefore crucial for the timely diagnosis and treatment of cancers.
We hypothesized that pillar[n]arenes could be used as biosensors for metabolites like 1-MNA. Pillar[n]arenes are pillar-shaped macrocyclic compounds with a polygonal cross-section (pentagonal and hexagonal for n = 5 and 6, respectively). We found that pillar[6]arene (P6A) forms host¬¬–guest a complex with 1-MNA; the metabolite can bind to it because the hexagonal cavity inside P6A offers just the right environment for doing so. We also found that when 1-MNA is bound to P6A, the fluorescent response of the latter significantly decreases — an effect that can be exploited as an indicator for the presence or absence of 1-MNA (strong or weak fluorescent response, respectively). Importantly, we could show that the P6A fluorescence detection mechanism works for biological samples. Specifically, they were able to detect 1-MNA in urine, albeit with a low sensitivity. We conclude that additional experiments “will help to improve the sensitivity and specificity of the biosensors”, and that their work “should contribute to the development of low-cost, easy, and rapid methods for the detection of human metabolites for diagnosis”. Now, we have shown that pillar[6]arene can be used as biosensors for the metabolite 1-MNA — an important result given that detecting low-molecular-weight metabolites is challenging.
リソソームは脳腫瘍治療法の新たな標的である!Novel therapeutic strategy of malignant brain tumors by targeting lysosome.
Hegazy AM, Yamada D, et al., J. Biol. Chem, 2016, Vu TH et al, Can. Sci. 2018, Jing Y. et al, Can. Sci. 2022
私たちの研究室では,悪性化制御において重要な分子の同定および分子メカニズムの解明とともに,ハイスループットスクリーニングによる新規化合物の探索や既存薬スクリーニングを通して臨床応用を目指している。その目的のため,遺伝子改変動物を用いた腫瘍モデル,がん患者から摘出した腫瘍組織由来培養細胞に関する実験手法の確立にも取り組んでいる。
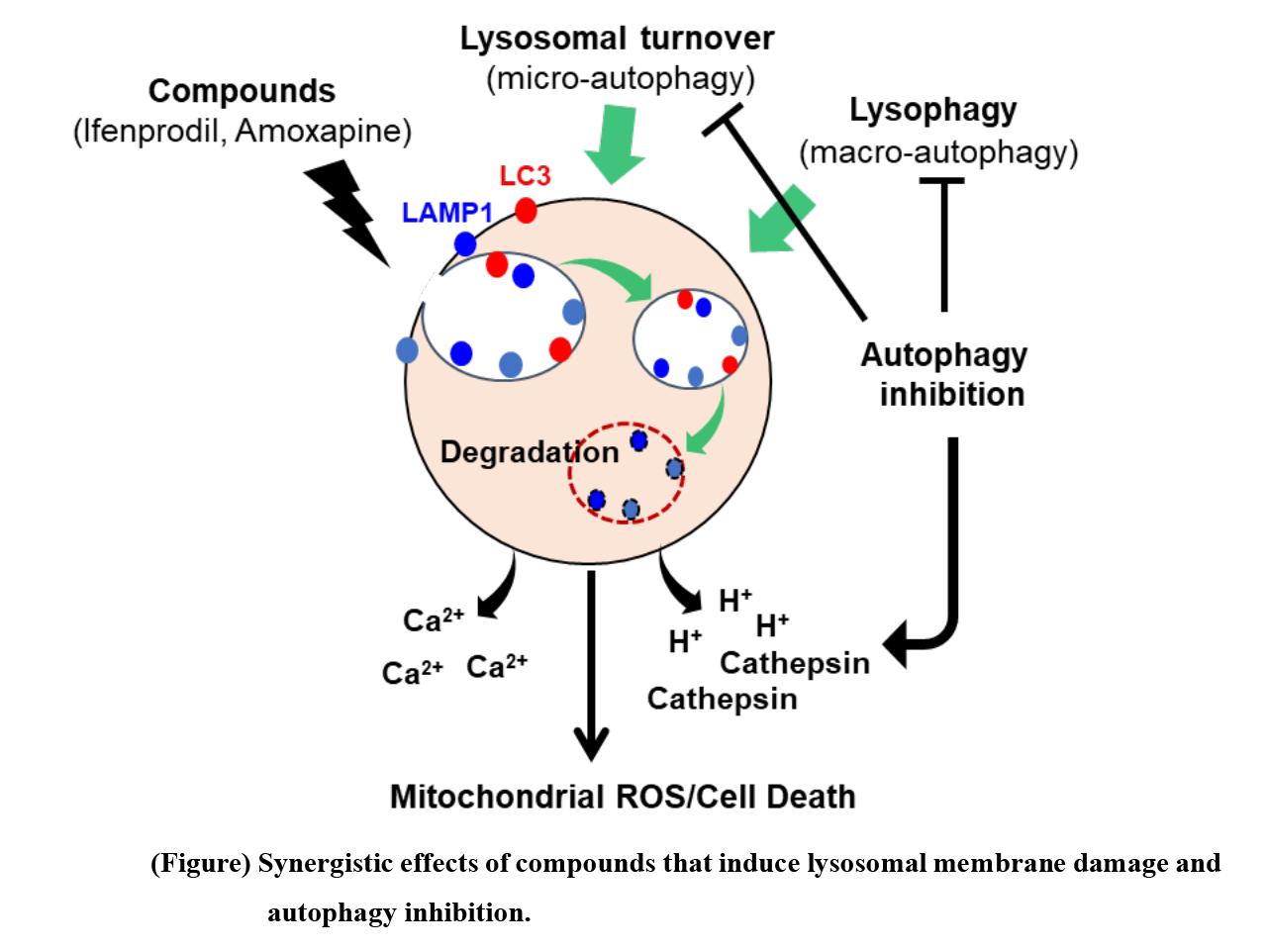
オートファジー・リソソームは,細胞内消化のプロセスを通じて,傷害を受けたオルガネラや蛋白質などの処理,再利用など,細胞の品質管理に寄与する。脳腫瘍に関しても,その生存に必須の役割を果たすという報告とともに,一方で相反するデータも報告されるなどい,一定の結論に至っていない。私たちは,遺伝子改変マウスを用いた動物モデル,さらには患者由来脳腫瘍細胞スフィア培養および同所移植(orthotopic PDX)を用いて,mTORおよびオートファジーの脳腫瘍における役割を解析した(Hegazy AM, Yamada D, et al., J. Biol. Chem, 2016. Vu TH et al, Can. Sci. 2018)。結果として,複数の患者由来脳腫瘍細胞においてはオートファジーを不活性化したにも関わらず,試験管内・生体内双方において大きな変化は認められなかった。しかし,更なる解析により,一部の細胞内カルシウム上昇を促す化合物群に対して感受性を示すことを見出した。これらの化合物とオートファジー阻害剤の組み合わせにより,顕著な抗腫瘍効果が発揮されることを見いだした。これらの化合物の作用機序を解析した結果,1.リソソーム膜透過性(LMP)を惹起する化合物がリソソーム由来のカルシウム上昇を促し,その後,ミトコンドリア由来活性酸素を上昇させ細胞死を誘導すること,2.リソソーム膜堅牢性は,オートファジー(Lysophagy)によって保たれていること、3.FDA承認薬の中でも、LMPを惹起する中枢神経系疾患薬(Ifenprodil)とオートファジー阻害剤(Chloroquine)を選択し、その併用がorthotopic PDXにおいても顕著な有効性を示すことを確認した。
一連の結果は、Lysosomeが有効な治療標的となることを示す知見である。今後,Lysosomeがいかにがん悪性化にとって重要なのか,またその制御メカニズムの詳細を解明することで,がんの悪性進展の理解と治療薬開発につなぐ研究展開を目指している。
We have attempted to develop novel treatment for malignant brain tumors (malignant gliomas) by identifying nutrient metabolic pathways that characterize malignant properties and searching for their inhibitors, focusing on mTOR, autophagy, and lysosome function, in patient-derived spheroid culture, patient-derived xenograft (PDX) and genetically engineered mouse models (GEM).
Firstly, we constructed a mouse model of malignant brain tumor in which mTORC1 is artificially activated by Tsc1 deficiency. By biochemical analysis, we observed that deletion of Tsc1 gene increased glioma-initiating cells (GIC), which contribute to glioma formation. Furthermore, mTORC1-activated glioma cells showed markedly increased ATP production along with enhanced glycolysis and oxygen consumption, suggesting that mTOR-dependent enhancement of energy production contributes to the amplification of GICs. Screening of compounds that exerted a strong inhibitory effect on Tsc1-deficient cells demonstrated the utility of targeting energy production in mitochondria(Hegazy AM, Yamada D, et al., J. Biol. Chem, 2016).
Secondly, we aimed to clarify the role of autophagy in malignant gliomas and attempted to disrupt the ATG5 gene using genome editing technology in patient-derived cells. We disrupted the ATG5 gene using CRISPR/Cas9 in several patient-derived cell lines and obtained defective clones; ATG5-deficient cells showed loss of autophagy activity, but no significant effects on cell growth, differentiation, or cell death. Furthermore, ATG5-deficient cells were not significantly different from wild-type cells when compared to their in vivo growth by intracerebral or subcutaneous transplantation into immunocompromised mice. In addition, we found that some compounds that induce autophagy, such as nigericin, showed strong antitumor effects against cells in combination with autophagy inhibition. Furthermore, we found that the combination of these compounds with autophagy inhibitors exerted a remarkable antitumor effect. Elevated cytosolic Ca concentration is known to impair mitochondrial function, and the compounds increased Ca concentration and directly impaired mitochondrial function, leading to loss of glioma cell undifferentiability (Vu TH et al, Can. Sci. 2018).
Thirdly, an FDA-approved existing drug screen was conducted to develop therapies targeting lysosomes. We found that drugs clinically used for CNS diseases, such as ifenprodil, were significantly effective to induce lysosome membrane permeability (LMP). Evaluation of Galectin3-GFP puncta formation in lysosomes confirmed the strong lysosomal membrane-disrupting effect of Ifenprodil. Furthermore, we found that these compounds induced an increase in intracellular calcium, followed by cell death due to increased ROS production in mitochondria. These phenomena were also inhibited by ROS scavenging (NAC), confirming that the compound can function as a therapeutic agent targeting the lysosomal-mitochondrial pathway. Its cell-cytotoxic effect of ifenprodil was increased when used in combination with autophagy inhibitors. Interestingly, they do not have a pronounced effect on normal neural progenitor cells, indicating that targeting lysosome functions is a novel therapeutic strategy of brain tumors (Jing YW. et al, 2022).
These results indicate that the lysosome is a unique and effective therapeutic target for glioma therapy. Based on the findings, we aim to elucidate how important lysosomal activity is for cancer malignancy and the details of their regulatory mechanisms, to understand the nature of malignant progression and to develop therapeutic agents.
mTORシグナルの抑制状態は,幹細胞特性の獲得を促すか? ~白血病幹細胞の自己複製と増殖におけるmTORシグナルの異なる役割~Does the suppressive state of mTOR signaling leads to the acquisition of stem cell characteristics?
Distinct roles of mTOR signals in regulation of self-renewal and propagation of leukemia cells.
Hoshii et al, J. Clin. Inv 2012, Hoshii et al., PNAS 2014
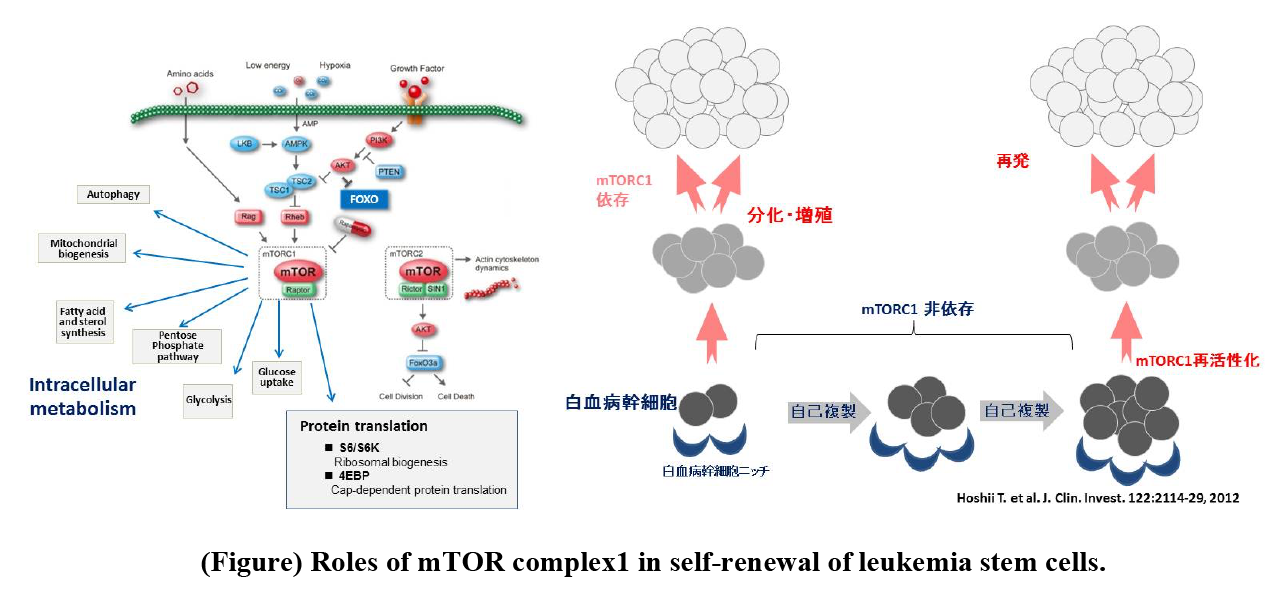
mTORは,Raptor,mLST8/GbL, PRAS40などからなるmTOR複合体1(mTORC1)とRictor, mLST8, SIN1, ProtorからなるmTOR複合体2(mTORC2)を構成するセリンスレオニンキナーゼである(図)。インスリン・成長因子-PI3K-AKT経路の活性化は,TSC2のリン酸化を誘導し,TSC複合体を不活化し,その標的であるRhebの活性化を通して,mTORC1の活性化を促す。また,Ragを介したアミノ酸による活性化,LKB1-AMPK経路による活性抑制など,様々な栄養シグナルのセンサーとして機能している。mTORC1の標的としての多数の分子が知られている。例えば,4E-BPは蛋白翻訳を促し,S6Kは,リボソームの生合成を促進することによって,細胞成長(細胞の大きさ)を制御する。また,mTORC1は,蛋白合成の促進だけではなく,解糖系などの糖代謝,脂質代謝,ペントースリン酸回路,オートファジー制御やミトコンドリアの代謝制御に関わる。一方、分子標的治療薬mTOR阻害剤Rapamycinは,FK506 binding protein 12 (FKBP12)と結合し,この複合体がmTORと結合することによって,mTORC1機能を阻害し,腎細胞がんや進行性膵内分泌腫瘍(pNET)など,一部のがんにおいて,臨床的有用性が確認されているなど、がんにおける重要なシグナルであることが知られている。
私たちは,がんにおけるmTORの役割を調べるために,薬剤(tamoxifen)誘導性Raptor欠損マウスを作成し,白血病モデルで解析を行った。その結果,Tリンパ球性白血病モデルにおいては,Raptor欠損により顕著な抗腫瘍効果を認めた(Hoshii et al., PNAS 2014)。一方で,急性骨髄性白血病モデルにおいては,白血病細胞全体としてはその増殖,生存が著しく障害され,個体レベルでの白血病の発症は顕著に抑制されていたが,一部の白血病細胞集団はmTORC1非依存的に長期的に生存していることが判明した。それらの集団は,白血病幹細胞と同様のマーカーを発現していること,Raptor遺伝子を再導入することにより,コントロールと同様の白血病形質に戻ることを観察した(Hoshii T, et al. J Clin Invest. 122:2114-29, 2012)(図)。これらの結果から,白血病幹細胞は,それ以外の白血病細胞と比較してmTORシグナルへの依存度が異なることが判明し,白血病幹細胞特有の細胞内代謝状態が存在することが示唆された。また,mTORC1シグナルを抑制すると,却って白血病幹細胞の数が増えるという現象も確認され,「mTORシグナルの抑制状態と幹細胞性」という新たな研究テーマを設定しそのメカニズム解明を進めている。
最近,Cell紙およびCancer Cell紙に相次いで「Diapause」と呼ばれる休眠状態ががんの治療耐性の鍵となることが示され,注目を浴びている(Cancer Cell 2021 39:142参照)。休眠状態のがんは,初期胚が環境変化に反応して,冬眠期間,発生プロセスを停止する現象(Embryonic Diapause)と共通分子基盤を背景とすること,具体的には,発がんを促進するmTORの活性を低下させると,休眠状態が誘導され,治療耐性の原因となるという,逆説的分子機構の存在が示された。この知見は,「mTORシグナルの抑制状態により幹細胞特性を獲得するという点で大変興味深い。
Mammalian/mechanical target of rapamycin (mTOR) is a highly conserved serine/threonine kinase in response to environmental determinants such as nutrient availability, energy sufficiency, stress, and growth factor concentration. mTOR forms two different complexes, called mTOR complex 1 (mTORC1) and mTORC2, and these complexes have distinct substrate molecules that function in the many biological processes. mTORC1 consists of several proteins, including an essential component, Raptor, contributes to promotion of anabolic pathways, i.e., enhancement of protein synthesis associated with increased energy production (Figure). Several clinical studies reported that high mTORC1 activity is co-related with poor prognosis of solid tumor patients, indicating that the anabolic processes promote malignant phenotypes of cancer cells. To investigate physiological roles of mTORC1 in leukemia cells in vivo, we developed several mouse leukemia models based on drug-inducible Raptor deficient mice. As a result, we revealed an indispensable role of mTORC1 in the development of T-cell leukemia (T-ALL) model driven by oncogenic Kras and Notch (Hoshii et al., PNAS 2014). In contrast, in the acute myeloid leukemia (AML) model, although Raptor deletion mainly induced apoptosis in majority of leukemia cells, LSCs survived and maintained in bone marrow long-term (Hoshii et al., JCI2012). The re-introduction of the Raptor gene into these Raptor-deficient AML cells induced them to propagate and differentiate. Thus, LSCs can survive under the mTORC1 inactivated condition, suggesting that they do not require the anabolic process for their self-renewal.
Recently, articles in Cell and Cancer Cell reported that a dormant state called "Diapause" has been shown to be a key factor in cancer resistance to treatment (see Cancer Cell 2021 39:142). Dormant cancers share a common molecular basis with embryonic diapause, a phenomenon in which an early embryo stops its developmental process during hibernation in response to environmental changes. The existence of a paradoxical molecular mechanism was demonstrated. This finding is very interesting in that the suppressive state of mTOR signaling leads to the acquisition of stem cell characteristics.
組織幹細胞および白血病幹細胞の静止期制御分子FOXOCritical roles of FOXO in dormancy of tissue stem cells and leukemia stem cells
Miyamoto et al, Cell Stem Cell 2007, Naka et al, Nature 2010
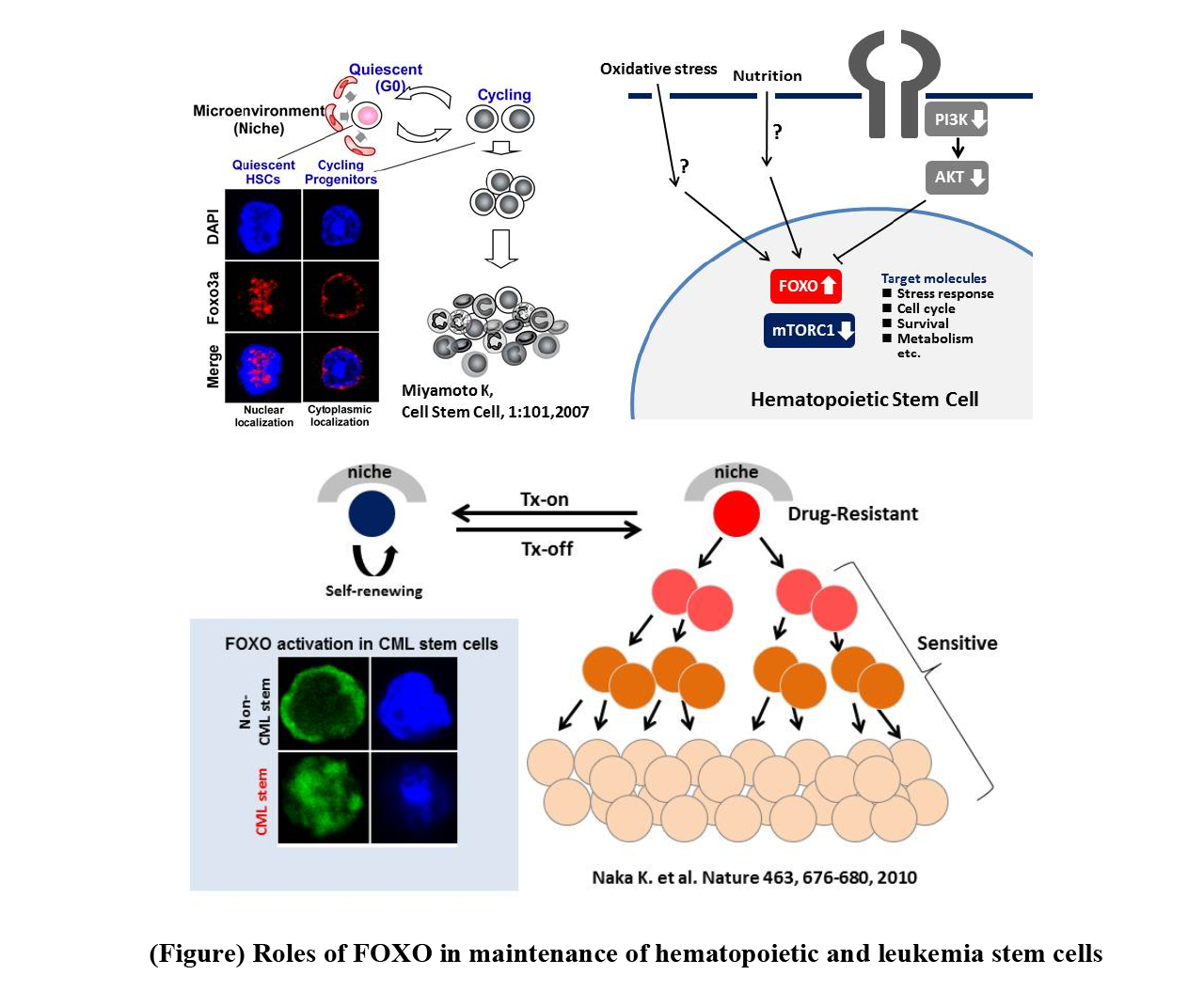
FoxOファミリーメンバーは,組織幹細胞に不可欠なシグナルと言える。哺乳類では,FoxO1,FoxO3,FoxO4,FoxO6がすべてPI3K-AKTシグナルの下流標的であり,成長因子やインスリンによる細胞刺激がない場合,FoxOは核に局在し,その転写標的を活性化する。成長因子やインスリンが適切な細胞表面受容体に結合すると,AKTが活性化され,FoxOsを直接リン酸化し,核から排除され細胞質で分解されることになる。代謝ストレスや酸化ストレスもまた,FoxOsの核内局在と転写活性を誘導することができる。
私たちを含む複数の研究グループが,FOXO遺伝子が,造血幹細胞の制御に深くかかわっていることを見出した(図)。静止期造血幹細胞では,FOXOは核内に局在していること,一方,前駆細胞に分化すると核外に移動することから,FOXOの挙動と未分化状態の関連が推察された。私たちは,FOXO3a欠損マウスの解析によって,この分子が造血幹細胞の骨髄再構築能,G0期維持,ストレス抵抗性に必須であることを示した(Miyamoto K, et.al. Cell Stem Cell. 1:101-112, 2007)。同様の結果は,Ghaffari らのグループからも報告されている(J Biol Chem 283:25692-25705, 2008)。一方,Gilliland らのグループは,FOXO1,3,4のトリプルコンディショナルノックアウトマウスの造血組織の解析により,単独の欠損に比べ,造血幹細胞,前駆細胞を含む未分化な細胞集団の著しい減少をきたすことを報告している(Cell 128:325-339, 2007)。これらの表現型は,抗酸化剤投与によって回復することから,活性酸素の上昇が造血幹細胞異常の原因であることが判明した。FOXOの転写因子としての直接の標的遺伝子としては,SOD2やカタラーゼなどの抗酸化酵素や細胞周期抑制因子(p57,p27)。これら一連の研究により,骨髄中のG0期の造血幹細胞におけるFOXOの活性化は,活性酸素レベルを低く保つことに重要であり,幹細胞の維持に必須の役割を果たしていることが明らかとなった(図)。最近,肥満マウスの造血幹細胞では,FoxOタンパク質はAKTなどの正常な上流制御因子に対して鈍感になり,高血糖がこれらの細胞でAKT-FoxO軸を直接変化させることが示唆された。このように,FoxOは,定常状態でも環境ストレス下でも,動物の健康な造血幹細胞を維持するために重要といえる。その他、神経幹細胞、筋肉幹細胞など、複数の組織幹細胞においてもその静止状態の維持に必須であり、組織横断的な幹細胞制御因子としてユニークな分子である。
私たちは,慢性骨髄性白血病幹細胞維持および治療抵抗性におけるFOXOの役割を解析した。マウス慢性骨髄性白血病モデルにおいて,白血病幹細胞集団(LIC)では,白血病前駆細胞とは対照的に,AKTの活性が低下していることを観察した。それに伴い,FOXOの核局在を示す細胞が多数観察された。さらに,FOXO3a欠損は,イマチニブによる治療効果の向上がみられ,本分子が白血病幹細胞の治療抵抗性に寄与していることを明らかにした(Naka K, et al. Nature 463:676-80, 2010 )。この結果は,pro-apoptotic tumor suppressorとして知られるFOXOが,腫瘍組織においてはその幹細胞性維持に寄与しているという点で,パラドックス的な現象であり,本分子の多面性を示した知見である。現在,私たちは,FOXO特異的阻害剤の開発を通じて,あらたな白血病治療薬の開発に取り組んでいる。
FoxO family members are vital for catabolic regulation in tissue stem cells. In C. elegans, reduced insulin/IGF-1 signaling extends lifespan in a process mediated by DAF16 (FoxO homolog). DAF16 and HLH30 (TFEB homolog) cooperatively regulate genes related to aging, protein homeostasis, and stress resistance, helping to prolong lifespan in a harsh environment. In mammals, FoxO1, FoxO3, FoxO4 and FoxO6 are all downstream targets of PI3K-AKT signaling. In the absence of cellular stimulation by growth factors or insulin, FoxOs localize in the nucleus and activate their transcriptional targets. When a growth factor or insulin binds to the appropriate cell surface receptor, AKT is activated and directly phosphorylates FoxOs, resulting in their nuclear exclusion and degradation in the cytoplasm. Metabolic or oxidative stress can also induce nuclear localization of FoxOs and their transcriptional activity. In HSCs, it is FoxO1 and FoxO3 that are mainly expressed and localized in the nucleus under conditions of AKT inactivation.
Several research groups, including ours, have found that the FOXO gene is closely involved in the regulation of HSCs. In quiescent HSCs, FOXO is localized in the nucleus, whereas it migrates out of the nucleus upon differentiation into progenitor cells, suggesting a link between FOXO behavior and the undifferentiated state. By analyzing FOXO3a-deficient mice, we showed that this molecule is essential for the bone marrow remodeling ability, G0 phase maintenance, and stress resistance of HSCs (Miyamoto K, et.al. Cell Stem Cell. 1:101-112, 2007). Similar results were reported by Ghaffari et al. (J Biol Chem 283:25692-25705, 2008). On the other hand, Gilliland et al. reported that analysis of hematopoietic tissues from triple-conditional knockout mice for FOXO1,3,4 resulted in a marked reduction in the undifferentiated cell population, including hematopoietic stem and progenitor cells, compared to the single deletion (Cell 128:325-339, 2007). These phenotypes were restored by antioxidant administration, indicating that elevated ROS is the cause of HSC abnormalities; direct target genes for FOXO as a transcription factor include antioxidant enzymes such as SOD2 and catalase, and cell cycle inhibitors (p57,p27). These series of studies revealed that activation of FOXO in G0-phase HSCs in the bone marrow is important for keeping ROS levels low and plays an essential role in stem cell maintenance (Figure). In HSCs from obese mice, FoxO proteins become insensitive to their normal upstream regulators such as AKT, suggesting that hyperglycemia can directly alter the AKT-FoxO axis in these cells. Thus, FoxOs are important for the maintenance of healthy HSCs in animals both at steady-state and in those under environmental stress. In NSCs, loss of FoxO1/3/4 or FoxO3 leads to defects in quiescence and undifferentiated status. Mechanistically, FoxOs control genes regulating the cell cycling and redox state of NSCs. In MuSCs, FoxOs are again critical for quiescence and prevention of premature aging. An intriguing study has revealed how FoxOs act to prevent of premature aging of MuSCs. Extremely old mice lose “genuine” MuSCs and gain “primed” MuSCs. FoxO1/3/4 deficiency causes a similar loss of “genuine” MuSCs, causing regenerative failure. Thus, FOXOs are unique molecules as stem cell regulators essential for the maintenance of quiescent status in several other tissue stem cells, such as neural and muscle stem cells.
Chronic myelogenous leukemia is caused by the Philadelphia (Ph) chromosome resulting from reciprocal translocations and the subsequent appearance of the BCR-ABL1 fusion gene, which has homeostatic tyrosine kinase activity and activates a variety of downstream molecules leading to leukemogenesis. The therapeutic agent imatinib, which selectively inhibits BCR-ABL1 tyrosine kinase, was developed as a treatment and has demonstrated remarkable therapeutic efficacy compared to conventional therapies. Recently, it was shown that tyrosine kinase inhibitors can inactivate BCR-ABL1 in leukemia stem cells, yet leukemia stem cells can survive and cause relapse. Interestingly, leukemic stem cells in the BCR-ABL1-inactivated state behaved like normal stem cells and appeared to self-renew. Thus, because chronic myeloid leukemia stem cells resemble hematopoietic stem cells, we analyzed the role of FOXO in chronic myeloid leukemia stem cell maintenance and resistance to therapy. In a mouse model of chronic myeloid leukemia, we observed that AKT activity is decreased in the leukemic stem cell population (LIC), in contrast to leukemic progenitor cells. Correspondingly, we observed a large number of cells exhibiting nuclear localization of FOXO. Furthermore, FOXO3a deficiency was associated with enhanced therapeutic response to imatinib, indicating that this molecule contributes to the therapeutic resistance of leukemic stem cells (Naka K, et al. Nature 463:676-80, 2010). This result is a paradoxical phenomenon in that FOXO, a known pro-apoptotic tumor suppressor, contributes to the maintenance of stemness in tumor tissues, a finding that demonstrates the multifaceted nature of this molecule. We are currently working on the development of new leukemia drugs through the development of FOXO-specific inhibitors.